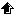
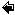
| | 13.5 Transformation of phosphorus in sediments |
The main transport mechanism of P to sediments is clearly the settling of particulate matter, although influx of dissolved P to sediment may also occur. The surface of the sediments receives a mixture of particulate inorganic and organic compounds containing P. In sediments, the settled particulate inorganic P can be divided into Fe-, Mn-, Al- and Ca-bound P, whereas the particulate organic P consists of living and dead algae, plant debris, zooplankton, bacteria and detritus. Part of the settled particulate P behaves as inert material and is buried in its original form, whereas part of the P (i.e. mobile P) is involved in various physico-chemical and biological reactions before final burial as inert material in sediments. Mobile pools of loosely sorbed P, Fe-bound P and fresh organic P may constitute over 50% of the surface TP but are largely depleted below a depth of couple of centimeters. The nonmobilized, buried P consists mostly of stable minerals such as apatite and refractory organic P, whereas the Fe-bound P constitutes only a minor proportion of the burial flux of P.
Particulate organic P is considered to be the main compound transporting P to sediments. In marine regions with marked seasonality such as at northern temperate latitudes, the settling of the spring bloom with its high P content carries a large portion of the organic particulate P to the surface of the sediment. In the Baltic Sea, for example, seasonal short-term changes in the phytoplankton biomass in the water mass are due to the rapid sedimentation of phytoplankton [225]. Large part of dissolved phosphorus may be stripped from the entire water column by this event. Although organic matter is mineralized during sinking, a substantial portion of the settled particulate organic matter decomposes within the sediments in relatively shallow environments. In estuaries, however, the sedimentation of particulate matter may be accentuated by salt-induced flocculation of colloidal and particulate matter containing P [142]; further, the dissolved P may precipitate on Fe(III) oxides [204]. The precipitation on Fe(III) oxides in water may be major binding process of P in water depths, where biological uptake is minor.
Labile organic matter rich in P tends to increase the release of P from sediment to water within a period of days after settling. The settling and subsequent mineralization of labile organic matter does not, however, necessarily lead to the immediate release of P from sediment to water owing to the sorption capacity of P in sediments. A major portion of the organic P in sedimentation flux can be mineralized in sediments, but the mineralized P is partitioned between the pore water and adsorption sites of the sediment. The proportion of organic P may then decrease but that of Fe(III) oxide-bound P increase in surface sediment. In Århus Bay, a large part of the P precipitated in spring is mineralized at the sediment surface and subsequently retained in the pool of Fe-bound P [250]. Efficient release of Fe(III) oxide-bound P has nonetheless been observed later, that is, in August. In the study of Sundby et al. [448], the settling matter had a much higher proportion of organic P (35% of TP) and a lower proportion of P bound to metal oxides (25% of TP) than was measured on the sediment surface (6% and 50% of TP, respectively).
Therefore, although the settling of particulate organic P is an important transport mechanism of P into sediment, the P is partly bound to inorganic compounds after mineralization in sediments. However, neither fresh organic P nor Fe(III) oxide bound P is efficiently buried, because organic P is mineralized and Fe-bound P is dissolved in the reduction of Fe(III) oxides in sediments [250]. In the Gulf of St. Lawrence, for example, approximately half of the sedimentation flux of particulate P is released from sediment back to water [448], whereas in Århus Bay, 65% of the net sedimentation of P is released back to water [250].
When does the release of P occur? In eutrophic lakes and marine coastal areas at northern temperate latitudes the increase of P concentration in near-bottom water occurs usually during summer and autumn, when the external P load is low due to the low river discharges. For example, in shallow lakes of Denmark, there is a clear seasonal pattern in the concentrations of P in water column in the eutrophic lakes, whereas no similar pattern is observed in the oligotrophic ones (Figure 4). The increase in P concentration can be explained with benthic P efflux. In the coastal Gulf of Finland, Baltic sea, the P concentration increases in the near-bottom water and the increase can be explained with enhanced benthic P efflux (Figure 4).
Several coinciding factors may explain the increase in benthic flux of P in mid-summer and early autumn. The settling of the phytoplankton serves as a fresh energy source for microorganisms, which in turn enhances microbial activity in sediments in summer. Seasonal changes in hydrographic features may also increase the benthic flux during the summer period. In summer, density stratification is strengthened due to the formation of the thermocline. Density stratification prevents the efficient mixing of surface and deep water layers, resulting in a prolonged residence time of the near-bottom water. Owing to this prolonged residence time and the high sediment O2 consumption caused by a high sediment organic matter content, the near-bottom O2 concentration decreases in summer. Although O2 is not depleted from the near-bottom water, the lowered O2 concentration may cause anoxia at the sediment-water interface, thus favouring the release of P. It is also likely that anoxia in sediment in summer permits anaerobic microbial Fe(III) oxide and SO42- reduction close to the sediment-water interface, which in turn affects the cycling of Fe, S and P in surface sediments.
Heterotrophic microbial reduction of electron acceptors during mineralization is considered to be a controlling factor in sediment P cycling ([43], see Section 12.3). The redox potential in sediment differs drastically from that in the oxic water mass, which affects P equilibrium between the solid and solution phases, i.e. sorption capacity of sediment. Thus, the transformation of previously unavailable forms of P into available ones may necessitate a temporary benthic step.
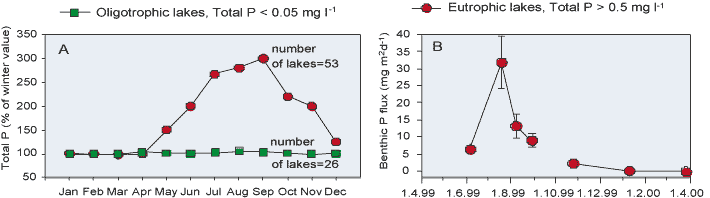
Heterotrophic microorganisms that reduce oxidized compounds using organic matter as an energy source are directly or indirectly responsible for most of the redox reactions in sediments (see Section 12.3). Phosphorus dissolved in pore water originates mainly from both Fe(III) oxides and organic matter, which constitute the main mobile P-binding pools in sediments. The bulk of the P, however, is thought to be released in sediment from Fe(III) oxides undergoing reduction. There are two main processes that can reduce Fe(III) oxides in sediments: first, enzymatic microbial dissimilatory reduction and, second, nonenzymatic reduction by the sulphide formed in microbial dissimilatory SO42- reduction [292].
Microorganisms are important in the reduction of Fe(III) oxides, because the storage under anaerobic conditions of readily reducible, synthetic, poorly crystalline Fe(III) oxides or sterilized sediment containing Fe(III) oxides does not result in detectable Fe(III) reduction, even after long periods. As well as anaerobic conditions, the Fe(III) oxide-reducing bacteria need direct contact with particles to reduce Fe(III). When direct contact is prevented, Fe(III) oxides are not reduced [292]. In microbial dissimilatory Fe(III) oxide reduction, Fe(III) is used as an external electron acceptor and Fe(III) is reduced to soluble Fe(II), organic C being the electron donor. Dissimilatory microbial Fe(III) reduction leads to the simultaneous accumulation of Fe(II) and P in anoxic pore water.
Although the concentration of O2 in near-bottom water is high, the anoxic zone develops within a few millimetres beneath the sediment-water interface in organic-rich, fine-grained marine sediments. O2 may be depleted, but the nitrate (NO3-) present can still inhibit Fe(III) reduction in sediments and so prevent the release of P from Fe(III) oxides to anoxic water [7]. Extensive release of P may occur after depletion of O2 due to the fact that nitrification is inhibited after the exhaustion of O2, leading rapidly to depletion of nitrate.
In marine systems, Fe(III) oxides are probably reduced mostly by H2S, because SO42- reduction is the dominant anaerobic respiration process (see Section 12.3). In Århus Bay, for example, partial oxidation of H2S accounts for 63% of the estimated Fe(III) reduction, whereas the contributions of microbial Fe reduction to carbon mineralization are considered to be small [461]. The rate and extent of microbial Fe(III) oxide reduction is controlled by the surface area and site concentration of the solid phase. Microbial reduction of poorly crystalline Fe(III) oxide is about 20 times that of goethite, which is about 50 times that of hematite [395]. This result is consistent with the decrease in particle size and increase in surface area and crystallinity of these oxides. In accordance with microbial Fe(III) oxide reduction, poorly crystalline Fe(III) oxide minerals are more reactive towards sulphide than are crystalline goethite and hematite. Thus, Fe(III) oxides that effectively adsorb P are the same forms as those that are effectively reduced by Fe(III) oxidereducing microorganisms and by H2S formed in microbial SO42- reduction in sediments.
A classic P cycling model is based on the Fe cycle in sediment: insoluble Fe(III) oxides are reduced to soluble Fe(II) ions, after which the P bound to Fe(III) oxides is released into pore water. When reduced soluble Fe(II) is diffused to an oxic environment (sediment surface or near-bottom water) it is oxidized to Fe(III) oxides having a high capacity to sorb P. Hence, sediments overlain by aerobic waters often have an Fe(III) oxide-rich surface layer. The precipitated Fe(III) oxides present in the surface layer are considered to effectively prevent P from entering the euphotic surface of water. Adsorption of P onto Fe(III) oxides may occur rapidly, because Fe(II) is oxidized within minutes or hours in the presence of O2. As well as chemically, Fe(II) is oxidized by lithotrophic Fe(II) oxidizing bacteria in surficial sediments [434]. Due to the cycling of Fe in sediments, P can be desorbed and adsorbed several times before its permanent burial or release to water. The P-rich surface layer is in a dynamic state and over time moves upwards as new sediment accumulate.
However, back in 1948, Hasler and Einsele suggested that SO42- increases the availability of P, a hypothesis that was later substantiated by Sugawara et al. [446]. Recent studies have also revealed the tight coupling between Fe, S and P in marine sediments [395][400]. The coupling of Fe with S leads to differences in P cycling between SO42--poor and SO42--rich systems (Figure 5). As pointed out earlier, SO42- reduction does not itself release P, but it indirectly promotes P mobilization and reduces the P retention capacity. SO42- has a double effect on the P cycle via reactions with Fe: first, Fe(III) oxides are effectively reduced by sulphide formed in microbial SO42- reduction and, ultimately, even very stable crystalline Fe(III) oxides are converted to FeS2 in sulphidic sediments [70]; second, dissolved Fe(II) is quickly and effectively removed from pore waters by the precipitation of solid FeS, and the formation of FeS2 in the presence of H2S formed in SO42- reduction. Co-precipitation of Fe and S leads to permanent burial of FeS minerals [70]. The FeS minerals formed adsorb P poorly at neutral pH. As a consequence, the upward flux of dissolved Fe(II) is decreased, thereby reducing or preventing the reformation of an Fe(III) oxide-rich surface layer able to adsorb P efficiently. However, the P solubilized from Fe(III) oxides is maintained in pore water.
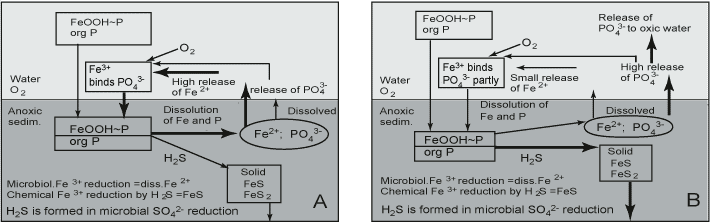
Due to FeS formation, the dissolved Fe:P ratio is higher in freshwater than in brackish-marine systems under anoxic conditions (Gunnars and Blomqvist 1997). When conditions turn oxic, the newly formed colloidal Fe(III) oxides scavenge P ions in lake-water sediment. For example, in Finland several hundreds of small lakes suffered from oxygen deficiency during winter 2002 - 2003. In these lakes both Fe and P concentrations increased in the whole water column, but after the ice melt and the increase of oxygen concentration in water both Fe and P concentrations went down. In marine sediments, though, P is less efficiently scavenged, and significant amounts remain dissolved in the overlying water (Gunnars and Blomqvist 1997). It has been suggested that the molar ratio of Fe:P dissolved in pore water governs the fate of P when these elements are transported to the oxic zone in sediment. The predominant Fe:P surface complexation ratio of Fe(III) oxide is suggested to be 2 [204](Gunnars and Blomqvist 1997). When the dissolved Fe:P molar ratio is under 2, the available Fe is in too short supply to bind all the dissolved P. In marine systems, the Fe:P ratio is under 2 and in lake-water systems (calcareous lakes not included) almost invariably above 2 under anoxic conditions (review by Gunnars and Blomqvist 1997).
The benthic flux of P increases when conditions turn from oxic to anoxic in lake (e.g. [147][323][324]), brackish-marine (Gunnars and Blomqvist 1997) and marine systems (e.g. [7][33]). Several studies have, however, confirmed the occurrence of significant aerobic P release in both lake and marine systems (e.g. [37][49][423][106]). In SO42- rich systems, a low concentration of dissolved Fe(II) may lead to insufficient Fe(III) oxide formation. The Fe(III) oxides formed can then only partially retain the flux of P, and the benthic efflux of P may occur even though the overlying water is oxic. In SO42- poor lakes, however, a portion of the P released to oxic water may originate from P bound to organic matter in sediments.
Owing to the low reserves relative to the requirements of algae in most lakes, P is a key factor in lake-water eutrophication [485]. In marine and brackish systems, including the Gulf of Finland, however, primary production in the photic zone tends to be N limited [222]. The difference in nutrient limitation can be partly attributed to the lower sediment P-binding ability due to the higher SO42- concentrations in brackish and marine sediments than in lake waters [395].
| | 13.5 Transformation of phosphorus in sediments |